Mapping with bwa¶
One of the most common operations when dealing with NGS data is mapping sequences to other sequences, and most commonly, mapping reads to a reference. Because short read alignment is so common, there is a deluge of programs available for doing it, and it is well-understood problem. Most of the efficient programs work by first building an index of the sequence that will be mapped to, which allows for extremely fast lookups, and then running a separate module which uses that index to align short sequences against the reference. Alignment is used for visualization, coverage estimation, SNP calling, expression analysis, and a slew of other problems. For a high-level overview, try this NCBI review.
Getting the Dependencies¶
bwa is one of the many available read mappers. bwa is a good “reference” version of a common alignment algorithm based on the Burrows-Wheeler transform; those of you looking to dive into a relatively thick paper on it can read about it here.
Firstly, let’s download bwa:
cd /root
curl -L "http://downloads.sourceforge.net/project/bio-bwa/bwa-0.7.5a.tar.bz2?r=http%3A%2F%2Fsourceforge.net%2Fprojects%2Fbio-bwa%2Ffiles%2F&ts=1379347638&use_mirror=softlayer-dal" > bwa-0.7.5a.tar.bz2
And then decompress it:
tar xjf bwa-0.7.5a.tar.bz2
Now, change into the directory with the source code and compile it:
cd bwa-0.7.5a/
make
Finally, move the compiled executable to a location where the operating system can find it:
cp bwa /usr/local/bin
So what did we actually just do? bwa is offered as an open-source program – that is, the actual code which defines its functionality is freely available. However, bwa is written in C, which, unlike Python, is not “interpreted.” Instead, the source code needs to be parsed and converted into machine language which can be run on the processor. This allows compiled programs to run much more efficiently than their interpreted counterparts, at the cost of needing to be recompiled for different systems. Although pre-compiled executables can be found for many programs, Unix programs are often distributed as source. ‘make’ is a program for tracking dependencies amongst files, and is used to manage the compilation of larger projects with many files.
‘make’ is also very useful for setting up pipelines, as it is not limited to source files – it can be used for any situation where one program needs to output of another to run. You can read about it here if you’re interested, though it’s outside the scope of this tutorial.
Getting the Data¶
We’ll be using the data we downloaded during the reads and quality control session; if you missed that, you’ll eventually want to run through it in Understanding Read Formats and Quality Controlling Data, but for now, you can just grab the quality-controlled data with:
cd /mnt
curl -O https://s3.amazonaws.com/public.ged.msu.edu/SRR390202.pe.qc.fq.gz
curl -O https://s3.amazonaws.com/public.ged.msu.edu/SRR390202.se.qc.fq.gz
You’ll also need a reference genome, which can be acquired with:
curl -O http://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/Escherichia_coli_O104_H4_2011C_3493_uid176127/NC_018658.fna
Mapping the Reads¶
To speed up the demonstration, we will just map a subset of the reads rather than the entire file, which is somewhat large (though small compared to many datasets). The head command outputs the first n lines of a file, by default 4:
cd /mnt/ecoli
gunzip -c ../SRR390202.pe.qc.fq.gz | head -400000 > ecoli_pe.fq
gunzip -c ../SRR390202.se.qc.fq.gz | head -400000 > ecoli_se.fq
We’ve got our reads and a reference, so we’re ready to get started. First, we build an index of the reference genome using bwa:
mv ../NC_018658.fna .
bwa index -a bwtsw NC_018658.fna
The -a flag tells bwa which indexing algorithm to use. The program will automatically output some files with set extensions, which the main alignment program knows the format of. Thus, we run the alignment like so:
bwa mem -p NC_018658.fna ecoli_pe.fq > aln.x.ecoli_NC_018658.sam
which aligns the left and right reads against the reference, and outputs them to the given SAM file. SAM is a common format for alignments which is understood by many programs, along with BAM. It’s often useful to have both, so we’ll use a utility called samtools to produce a sorted BAM file as well. First, install samtools:
cd /mnt
curl -O -L http://sourceforge.net/projects/samtools/files/samtools/0.1.18/samtools-0.1.18.tar.bz2
tar xvfj samtools-0.1.18.tar.bz2
cd samtools-0.1.18
make
cp samtools /usr/local/bin
cd misc/
cp *.pl maq2sam-long maq2sam-short md5fa md5sum-lite wgsim /usr/local/bin/
cd ..
cd bcftools
cp *.pl bcftools /usr/local/bin/
Then, run samtools to do the conversion:
cd /mnt/ecoli
samtools view -uS aln.x.ecoli_NC_018658.sam > aln.x.ecoli_NC_018658.bam
samtools sort aln.x.ecoli_NC_018658.bam aln.x.ecoli_NC_018658.bam.sorted
samtools index aln.x.ecoli_NC_018658.bam.sorted.bam
For additional resources on these tools, check out:
- the bwa manual
- info on samtools
- the SAM format spec
Visualizing your Data with Tablet¶
First, install dropbox using Installing Dropbox on your EC2 machine.
Now, copy your mapping files and the reference to your dropbox folder:
cp aln.x.ecoli_NC_018658.bam.sorted.bam* /root/Dropbox
cp NC_018658.fna /root/Dropbox
Although you can do many things with your alignments, one useful thing is to simply view them through a graphical interface. To demonstrate this, we’ll use a program called Tablet, which can be downloaded here.
Tablet claims to run on Windows, Linux, and OSX, though I have only tested it out on Linux. Because this is a GUI-driven program, we’ll be running it on our local machines instead of our EC2 instances. So, go ahead and grab the appropriate version for your system, and install it.
Once you have it installed, open it up. You’ll want to load your mapping file and reference genome:
You’ll then get some loading bars, and potentially an error about the indexing file which can be ignored. You need to select a contig on the left to view; ecoli has a very good reference, and is only one contig:
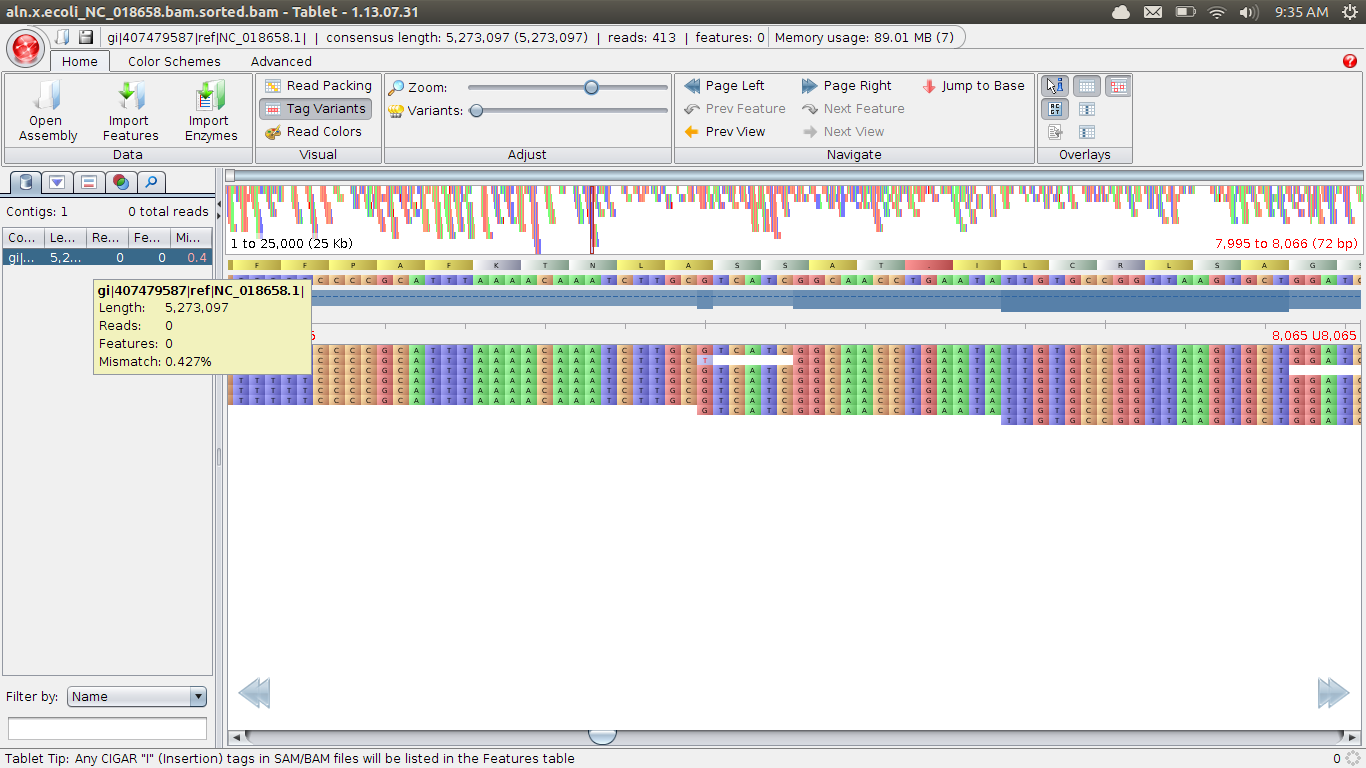
You can move left and right along the contig, as well as zoom. Tablet can view other information like gene structure, but we won’t get into that.